教学媒体资源服务平台
登录
课程视频库
北大讲座网
媒体编辑工具
教学备课资源
视频制作与支持
北京大学直播
人物专栏
教室资源
拍摄与直播服务
演播室预约
好学app下载
首页
讲座视频
讲座预告
讲座视频
推荐视频
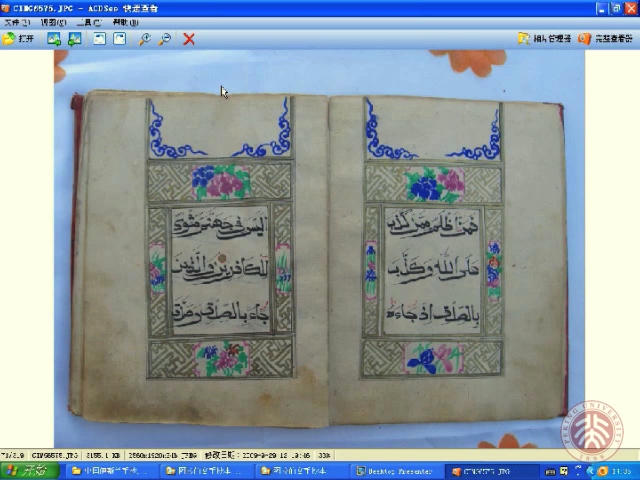
中国伊斯兰手抄本研究与伊斯兰手稿学
本讲座将围绕中国伊斯兰手抄本这一独特有趣的文化主题,论述中国伊斯兰手抄本在我国的产生和发展、基本特点、分布区域、语言种类、内容类别、装帧种类及艺术、彩绘种类、彩绘特点、书法艺术、搜集整理、抢救保护、手抄本的鉴定、数字化、出版以及研究现状,同时也简要论述作为学科的伊斯兰手稿学和国外学术界对伊斯兰手稿学研究的历史与现状。使我们从更广阔的文明角度,了解中国伊斯兰手抄本对回族穆斯林精神生活的巨大影响,见证中国伊斯兰文明与中华文明互动的历程,体验多元文化在我们现实生活中的意义。
主讲人:虎 隆 研究员
时间:2018/09/17
语言:中文
地址:
上古汉语的声调问题
主讲人:唐作藩先生
时间:2018/09/17
语言:中文
地址:
The Garden of Earthly Delights: A Triptych
The talk takes the work of the 15th century Flemish painter Hieronymus Bosch, The Garden of Earthly Delights, as a starting point to discuss several interesting points related to European History during the Renaissance, such as the medieval imagi
主讲人:Diego Loukota
时间:2018/09/17
语言:英语
地址:
General Education in American Higher Education
主讲人:Dr. Craig K. Pepin
时间:2018/09/17
语言:英语
地址:
中国司法改革的走向
【主办方】北京大学法学院人大与议会研究中心
主讲人:贺卫方 何 兵 徐 昕
时间:2018/09/17
语言:其他
地址:.
按发布时间排序
按点击量排序